Seleccione un país o region



-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)

-
Italia (Italiano)

-
Hungary (Magyar)

-
Lietuva (Lietuviškai)

-
Mexico (Español)
-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)

-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)

-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
Entender/genética de ame entienda qué causa la Atrofia Muscular Espinal (AME)
La atrofia muscular espinal es causada por una mutación en el gen de supervivencia de la neurona motora 1 (SMN1).
E
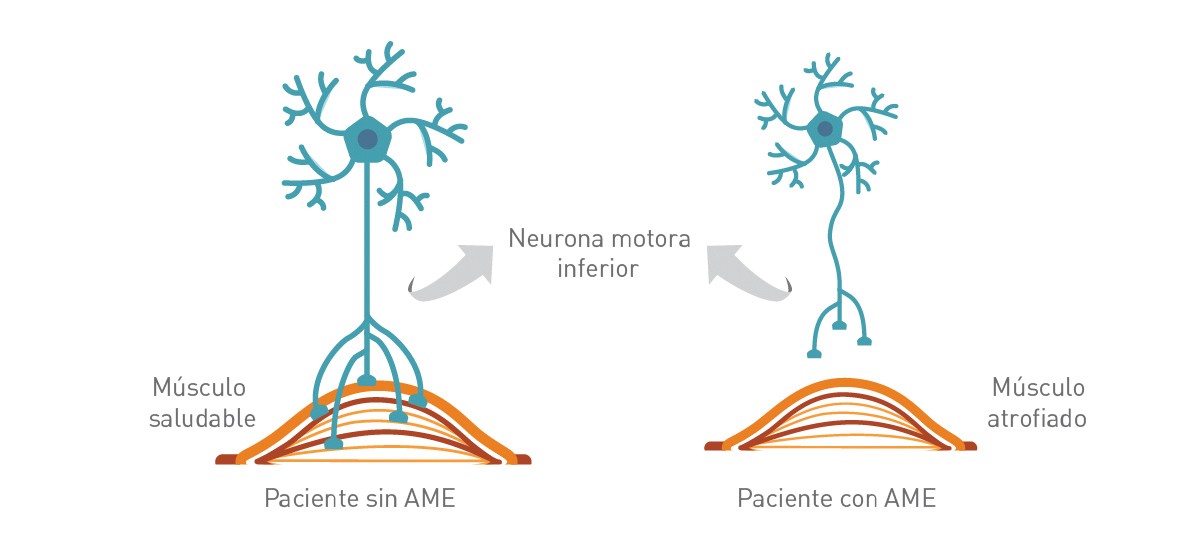
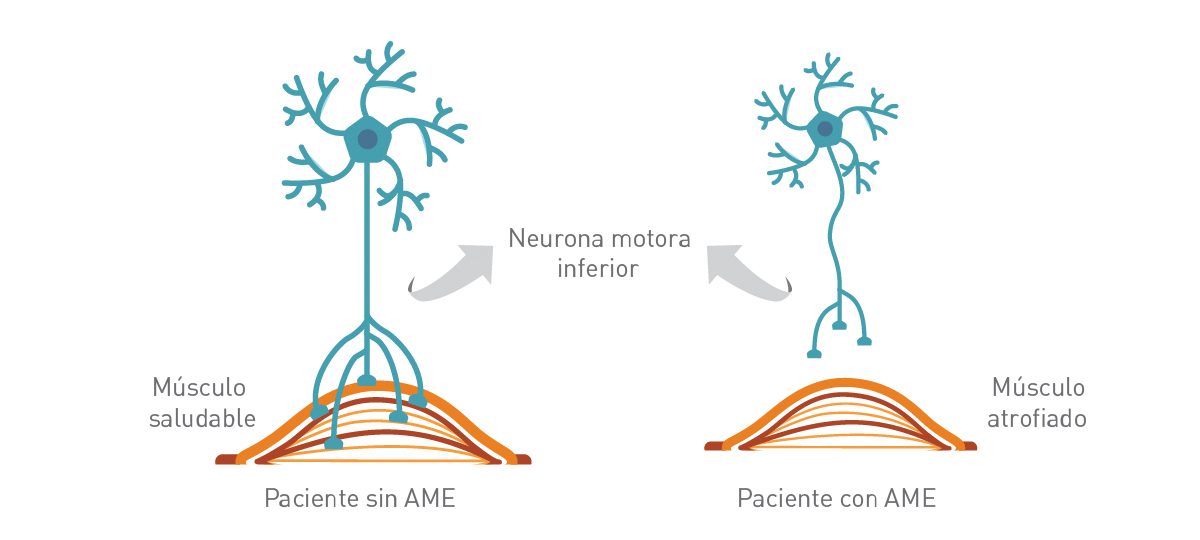
n la Atrofia Muscular Espinal (AME) hay una pérdida importante de células de la médula espinal - llamadas neuronas motoras - que son esenciales para controlar el movimiento y la fuerza muscular.
Estas neuronas motoras regulan la actividad muscular emitiendo señales desde el Sistema Nervioso Central (SNC), la parte del sistema nervioso que incluye al cerebro es la médula espinal.
La pérdida de funcionamiento de las neuronas motoras lleva a una debilidad y atrofia muscular progresiva (disminución gradual de la masa y la fuerza muscular) a medida que los músculos dejan de recibir señales del SNC.
En contraste con otras enfermedades neuromusculares raras, en la atrofia muscular espinal existe una clara comprensión de su origen genético.
¿Qué causa la Atrofia Muscular Espinal (AME)?
La atrofia muscular espinal es causada por una mutación en el gen de supervivencia de la neurona motora 1. Este gen es responsable de la producción de la proteína de supervivencia de la neurona motora (SMN), que mantiene la salud y función normal de las neuronas motoras.
En personas con AME, ambas copias del gen SMN1 presentan mutaciones, lo que dirige a una disminución de la producción de la proteína SMN. Sin un nivel adecuado de proteína SMN, las neuronas motoras de la médula espinal no funcionan correctamente, lo cual impide que los músculos reciban las señales adecuadas del cerebro.
La degeneración de las neuronas motoras lleva a una disminución gradual de la masa y fuerza muscular (atrofia).
¿Qué es la proteína SMN?
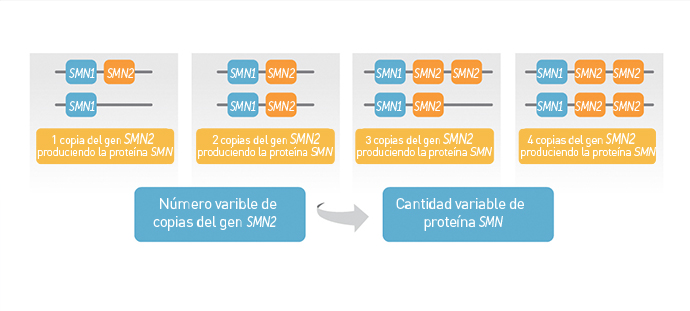
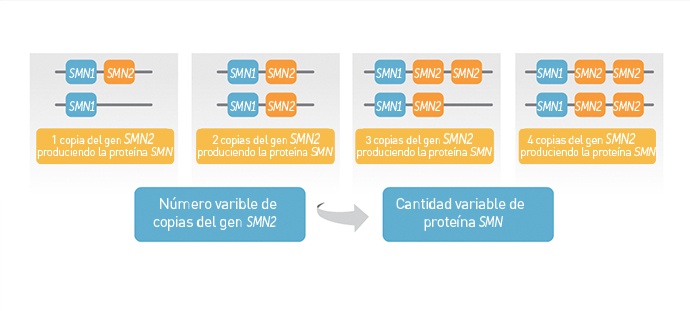
La proteína de supervivencia de las neuronas motoras (SMN por su siglas en inglés) se produce a partir del gen SMN1 que, en pacientes con AME, tiene alteraciones genéticas que impiden su función (1). La proteína SMN también puede producirse a partir de un segundo gen muy similar al SMN1, conocido como gen SMN2. Sin embargo, sólo una pequeña parte de la proteína SMN producida por el gen SMN2 es funcional y esta cantidad limitada no es suficiente para realizar el funcionamiento normal de las neuronas motoras (2).
El número de copias del gen SMN2 puede variar. Menos copias de SMN2 se asocian (generalmente) con síntomas menos severos de la atrofia muscular espinal. Aun así, la enfermedad tiene un amplio intervalo de síntomas y es difícil predecir su severidad solamente con el número de copias del gen SMN2. Por lo tanto, los expertos recomiendan que las decisiones sobre el cuidado integral se tomen con base en la capacidad funcional de cada paciente y no sólo en el número de copias de SMN2.
El descubrimiento del gen SMN2 ha proporcionado una oportunidad única para el desarrollo de terapias potenciales que pueden ayudar a este gen a producir más proteína SMN.
Referencias
1. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155-165.
2. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
¿Tiene dudas de algún término de este artículo? Revise el glosario.